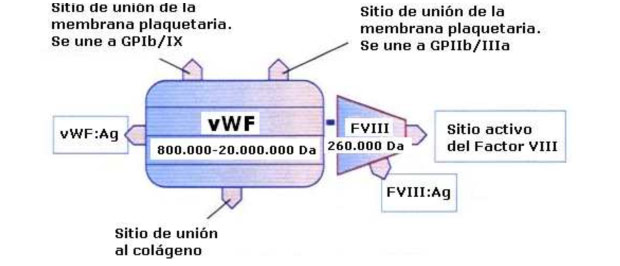
Figura 1. Complexo vWF/FVIII (Tomada de Rodak, 2005).
Aínda que o vWF actúa como transportador e estabilizador do Factor VIII, o seu principal función é participar na función plaquetaria concretamente na adhesión das plaquetas ás fibras de coláxeno que quedan ó descuberto tras unha ferida ou dano endotelial, feito que inicia unha cascada de interaccións entre as plaquetas, o vWF e as glucoproteínas Ib/IX y IIb/IIIa, ata unirse ó fibrinóxeno para mediar a agregación plaquetaria irreversible.
Fisiopatoloxía da enfermidade de von Willebrand. Tipos e subtipos
Os defectos, en xeral, na molécula do vWF tanto na estrutura como na función tradúcense nunha anomalía da adhesión plaquetaria que provoca unha hemorraxia sistémica e generalizada leve. Ademáis, a deficiencia cuantitativa de vWF produce unha deficiencia secundaria de Factor VIII xa que este faise máis vulnerable á degradación en plasma. Cando la deficiencia do vWF é grave os niveis de Factor VIII disminuen de tal forma que se produce un estado semellante ó da Hemofilia A grave e as hemorraxias son, ademáis de sistémicas, anatómicas e graves.
En función do tipo de mutación no xen que codifica o vWF pódese establecer unha clasificación cada vez máis precisa desta enfermidade. Así, na actualidade, a clasificación realízase en base á concentración plasmática do vWF, á actividade biolóxica e ós patróns dos multímeros (Figura 2).
Enfermidade de von Willebrand tipo 1
A deficiencia de producción do vWF é autosómica (non ligada ó sexo) dominante e polimorfa de orixe xenético. Os niveis de vWF e do Factor VIII son variables ainda que sempre se encontran diminuidos o que causa hemorraxia sistémica leve ou moderada. Esta é a forma más común e se observa en case o 80% dos afectados pola enfermidade de von Willebrand.
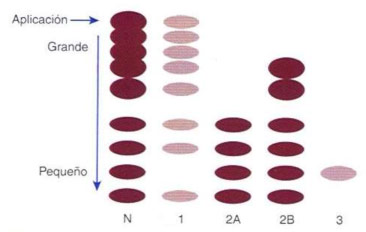
Figura 2. Análises do multímero del vWF por electrofreése. Diminución notipo 1, ausencia no tipo 2 e ausencia de case tódolos multímeros no tipo 3 (Tomada de Rodak, 2005).
Enfermidade de von Willebrand tipo 2
Hai distintos subtipos da Enfermidade de von Willebrand, que se explican a continuación:
Enfermidade de von Willebrand subtipo 2A
Dase no 10-20% dos pacientes con enfermidade de von Willebrand. Prodúcese por mutacións autosómicas dominantes que afectan o dominio estructural “A” da molécula do vWF que o fan máis sensible á degradación en plasma. A concentración del vWF é casi normal pero hai menos multímeros de alto e intermedio peso molecular que son os que participan máis íntimamente na agregación plaquetaria.
Enfermidade de von Willebrand subtipo 2B
Débese a mutacións que afectan ó dominio A1 que se traduce nun aumento da afinidade pola glucoproteína plaquetaria Ib/IX o que fai cos multímeros grandes se unan espontáneamente ás plaquetas e non estén disponibles para a adhesión plaquetaria normal.
Enfermidade de von Willebrand subtipo 2M
É unha variante cualitativa con diminución de interacción plaquetaria e ten un patrón multimérico normal. Confúndese no diagnóstico con el tipo 1.
Enfermidade de von Willebrand variante Normandía, subtipo 2N ou Hemofilia autosómica
Prodúcese a causa dunha mutación autosómica no xen do Factor VIII que codifica a rexión que se une ó vWF polo que se atopa afectada a súa estabilidade en plasma. Esta mutación, sen embargo, non afecta á función coagulante normal do Factor VIII. Cursa con hemorraxias anatómicas leves en homes e mulleres.
Enfermidade de von Willebrand tipo 3
Neste caso o vWF está ausente ou prácticamente ausente co que no se produce a estabilización do Factor VIII e polo tanto os síntomas son semellantes ós dunha Hemofilia A grave.
Outros factores condicionantes da enfermidade de von Willebrand
Existen outros factores que condicionan a aparición da enfermidade de von Willebrand a parte das mutacións xenéticas, tales como: o grupo sanguíneo ABO, as hormonas, o exercicio e o estrés determinan a producción do vWF no organismo. Os pacientes do grupo sanguíneo O teñen niveis máis baixos de vWF e unha incidencia maior que os de grupos A, B e AB. As elevacións de estróxenos durante o embarazo normalizan os niveis de vWF incluso nunha deficiencia moderada de vWF pero despois do parto os niveis caen bruscamente producíndose hemorraxias agudas. Tamén os niveis de vWF aumentan en procesos inflamatorios, co exercicio e co estrés, e en xeral os síntomas hemorráxicos diminuen coa idade.
Diagnóstico da enfermidade de von Willebrand
O diagnóstico é, en xeral, difícil xa que os distintos parámetros que se poden evaluar son altamente variables, incluso entre membros da mesma familia e co mesmo tipo de mutación.
A detección no laboratorio e a clasificación da enfermidade realízase fundamentalmente polas probas que se indican na Tabla 2: Tempo de sangría; cofactor ristocetina; antíxeno vWF; relación CBA/antíxeno vWF; reconto de plaquetas; TTPA; RIPA; actividade do Factor VIII e perfil de multímeros del vWF.
Tratamento da Enfermidade de von Willebrand
Xa que en xeral a enfermidade preséntase con manifestacións leves, o tratamento das hemorraxias locais palíanse con presión e compresas de xeo. Naquelas situación de hemorraxias empréganse estróxenos e DDAVP que liberan vWF dos sitios de almacenamento (Tabla 3). Na enfermidade de von Willebrand grave (subtipos 2A, 2B e tipo 3) empréganse concentrados de Factor VIII ricos en vWF obtidos de plasma humano. Na actualidade, estanse ensaiando con gran éxito concentrados de vWF recombinante que son máis seguros.
Tabla 2. Pruebas de laboratorio para la detección y clasificación de la enfermedad de von Willebrand
| Proba laboratorio |
Tipo 1 |
Subtipo 2A |
Subtipo 2B |
Tipo 3 |
| Tempo de sangría |
>10 minutos |
>10 minutos |
>10 minutos |
>10 minutos |
| Cofactor ristocetina |
>50% de actividade |
>50% de actividade |
>50% de actividade |
>10% de actividade |
| Antígeno vWF |
<50% |
<70% |
<70% |
<10% |
| Razón CBA/vWF:Ag |
>0,5 |
>0,5 |
<0,5 |
Non aplicable |
| Nº de plaquetas |
150.000-400.000/mL |
150.000-400.000/mL |
<150.000/mL |
150.000-400.000/mL |
| TTPA |
Algo prolongado |
Normal |
Normal |
Prolongado |
| RIPA |
Disminúe a agregación co uso de 1 mg/mL de ristocetina |
Disminúe a agregación co uso de 1 mg/mL de ristoceti |
Agregación co uso de 0,25 mg/mL de ristocetina |
Falta de agregación co uso de 1 mg/mL de ristocetina |
| Actividade Factor VIII |
30-50% |
<70% |
<70% |
<5% |
| Multímeros de vWF |
Concentración disminuida. Relación entre multímeros normal |
Concentración casi normal. Menos multímeros grandes e intermedios |
Concentración casi normal. Menos multímeros grandes |
Todos os multímeros están moi disminuidos |
CBA, ensaio de unión ó coláxeno; RIPA, agregometría plaquetaría inducida por ristocetina; TTPA, tempo de tromboplastina parcial activada.
| Tipo | Tratamiento primario | Tratamiento secundario |
| Tipo 1 | Estrógeno, DDAVP, EACA | Concentrado de Factor VIII |
| Subtipo 2A | Estrógeno, DDAVP, EACA | Concentrado de Factor VIII |
| Subtipo 2B | Concentrado de Factor VIII | |
| Tipo 3 | Concentrado de Factor VIII | Concentrado de plaquetas |
| Hemofilia autosómica | Concentrado de Factor VIII | |
DDAVP, acetato de desmopresina; EACA, ácido e-aminocaproíco.